

Understanding IPF
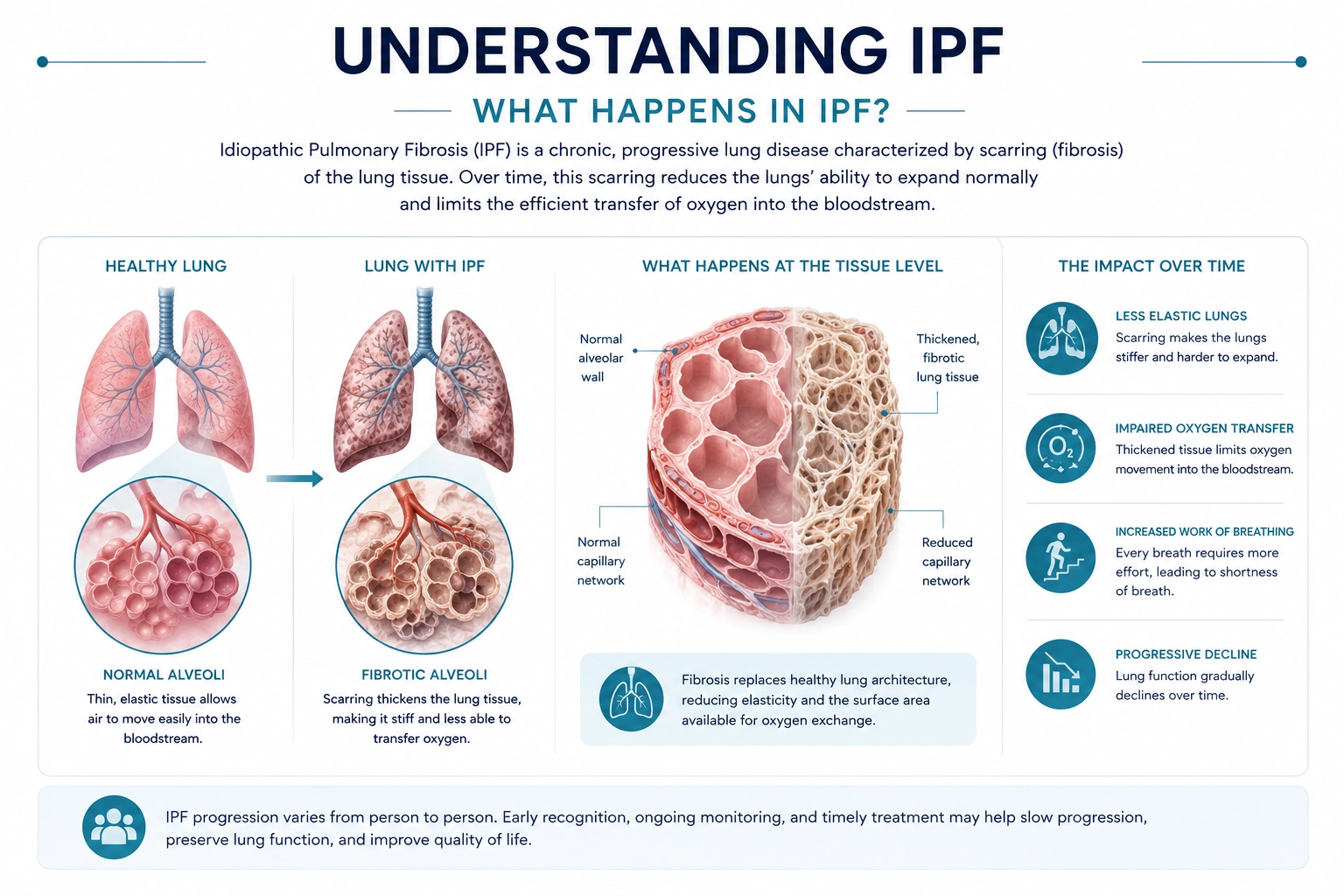
What Happens in IPF?
Healthy lungs are designed to stretch and allow oxygen to move efficiently into the bloodstream.
In IPF, progressive fibrosis causes the lungs to become thicker, stiffer, and less elastic, making breathing more difficult and reducing oxygen delivery throughout the body.
Over time, patients may experience:
- Increasing difficulty breathing
- Reduced exercise capacity
- Lower oxygen levels
- Progressive decline in lung function
Disease Progression
IPF progression varies from person to person.
Some individuals experience relatively gradual changes over years, while others may experience more rapid decline or episodes of sudden worsening known as acute exacerbations.
Disease progression may lead to:
- Increasing oxygen requirements
- Reduced mobility and activity tolerance
- Progressive decline in respiratory function
- Greater symptom burden over time
Early recognition and ongoing disease monitoring may support more informed treatment decisions.
Clinical Assessment and Monitoring
Diagnosis and monitoring of IPF often involve integrating clinical findings with imaging and pulmonary function testing.
Common tools used to support evaluation may include:
- High-resolution computed tomography (HRCT)
- Pulmonary function testing (PFT)
- Forced vital capacity (FVC)
- Diffusing capacity for carbon monoxide (DLCO)
- Oxygen saturation assessment
- Exercise testing (such as the 6-minute walk test)
These assessments may help characterize disease severity and monitor progression over time.
- Repeated microscopic injury to lung tissue
- Abnormal epithelial repair mechanisms
- Activation of fibroblasts and myofibroblasts
- Excess extracellular matrix deposition
- Pro-fibrotic signaling pathways (including TGF-β)
- Progressive remodeling of lung architecture
These mechanisms continue to be areas of active therapeutic investigation.
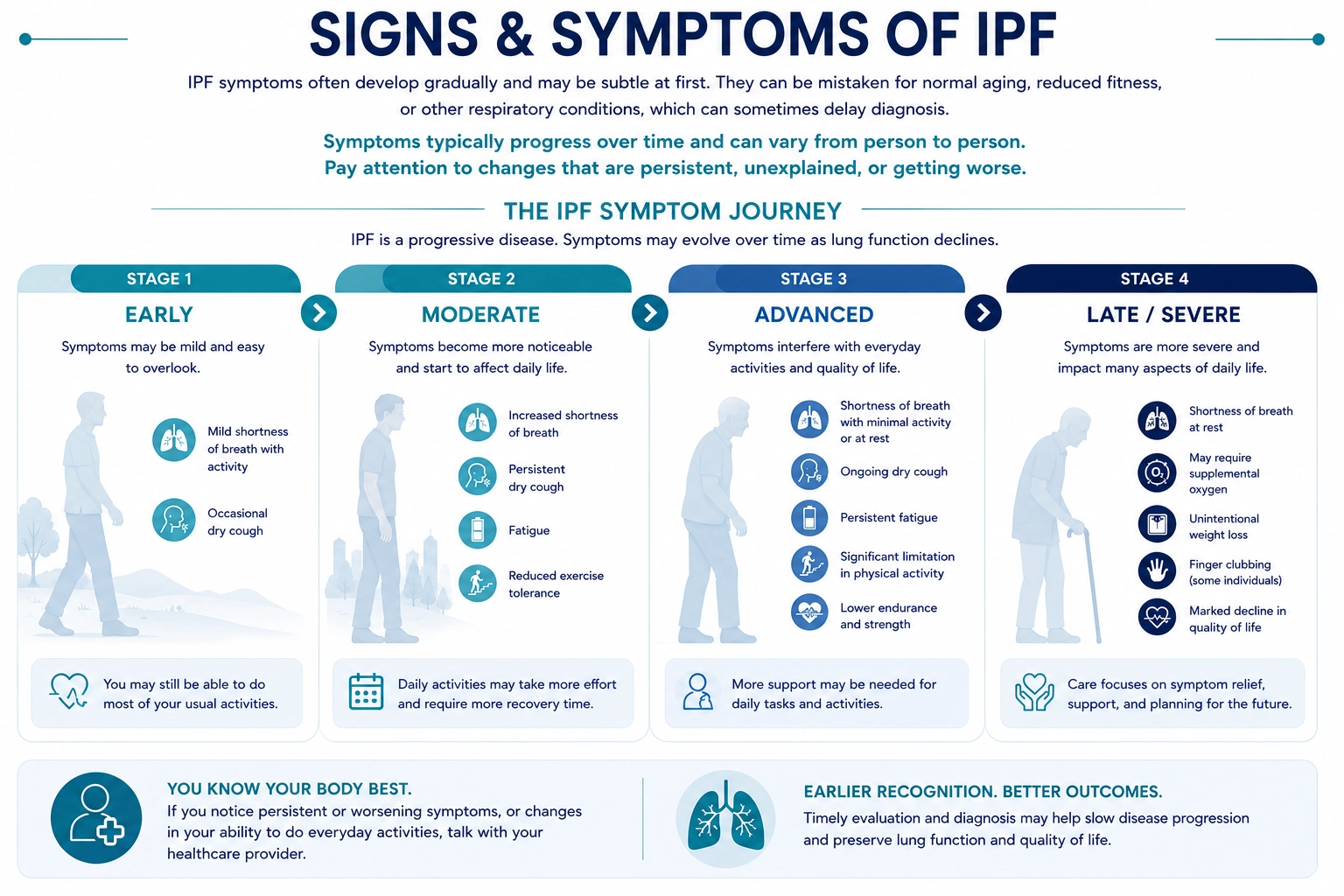
Signs & Symptoms
Idiopathic Pulmonary Fibrosis (IPF) often develops gradually, and early symptoms may be subtle or easily mistaken for normal aging, reduced fitness, or more common respiratory conditions. As a result, diagnosis may sometimes be delayed—particularly during the early stages of disease.
Symptoms can vary from person to person and may change over time as lung function becomes progressively affected. Rather than focusing on any one symptom in isolation, it is often more helpful to pay attention to changes that are persistent, unexplained, worsening over time, or beginning to interfere with normal daily activities.
The figure below highlights some of the more commonly recognized signs and symptoms associated with IPF and is intended to support awareness and encourage earlier evaluation.
Earlier recognition and evaluation may support more timely diagnosis and help preserve lung function and quality of life.
Risk Factors
Several factors have been associated with increased risk of developing IPF, including:
Current Treatment Approaches
Why Anti-Fibrotic Therapy?
Unlike some respiratory diseases that primarily involve inflammation, IPF is characterized by progressive fibrosis and abnormal wound repair. Current therapeutic approaches focus on slowing the progression of scarring, preserving lung function, and helping patients maintain quality of life.
Research continues to explore new approaches intended to target underlying fibrotic pathways and support more effective disease modification.
While there is currently no cure for IPF, treatment focuses on slowing disease progression, managing symptoms, and improving quality of life.
- May help reduce the rate of decline in lung function
- Most effective when started earlier in the disease course
- Regular monitoring is important to assess benefit and tolerability
- May be used during activity, sleep, or continuously
- Helps relieve shortness of breath and fatigue
- Titrated to individual needs and monitored over time

- Improves exercise capacity and endurance
- Teaches breathing techniques and energy conservation
- Provides emotional support and disease education

- Medications may help manage cough, reflux, or other associated symptoms
- Nutrition and weight management
- Psychosocial support and advanced care planning
- Evaluated at specialized transplant centers
- Careful selection based on overall health and criteria
- Post-transplant care includes lifelong follow-up and medications
